Two-electron SCF Fortran Code
Updated: 05 Aug 2025
Table of Content
- Table of Content
Main Program
two-electron SCF fortran code diagram
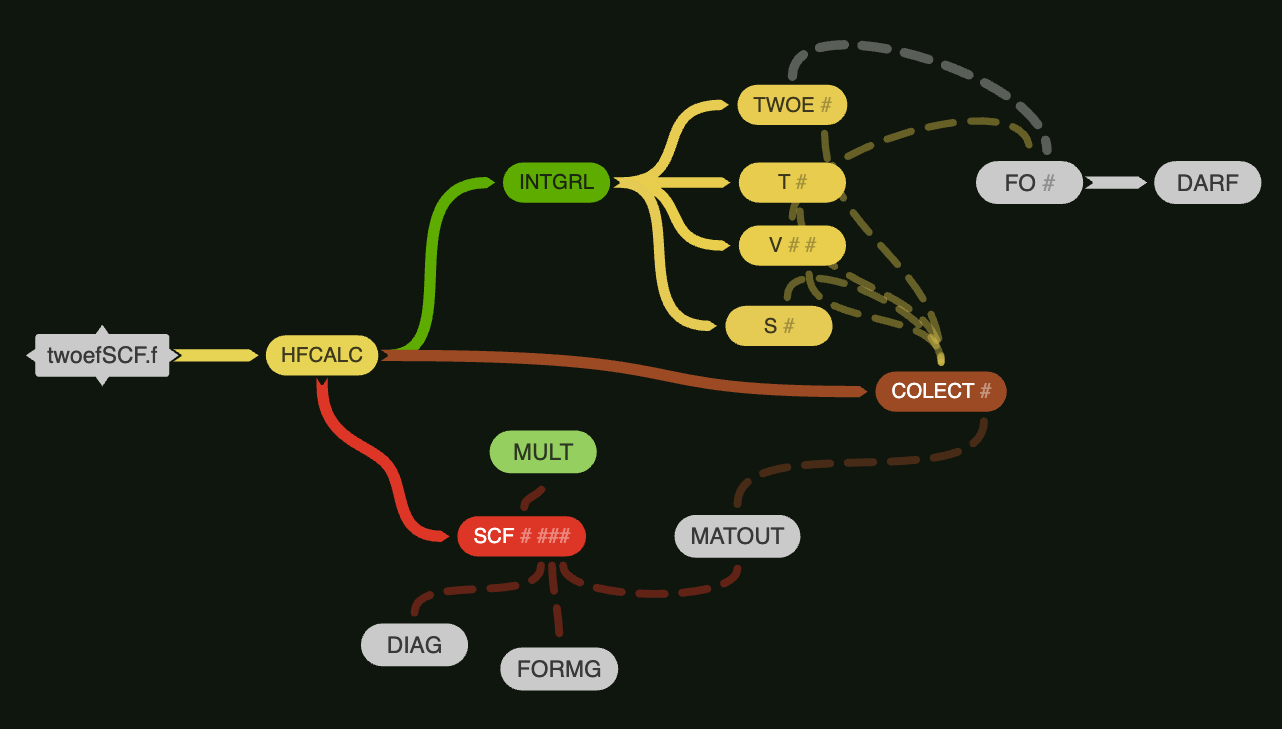
HF Calculation
IMPLICIT DOUBLE PRECISION(A-H, O-Z)
IOP = 2
N = 3
R = 1.4632D0
ZETA1 = 2.0925D0
ZETA2 = 1.24D0
ZA = 2.0D0
ZB = 1.0D0
! Call the subroutine to perform the calculation
CALL HFCALC(IOP, N, R, ZETA1, ZETA2, ZA, ZB)
END
SUBROUTINE HFCALC(IOP, N, R, ZETA1, ZETA2, ZA, ZB)
IMPLICIT DOUBLE PRECISION (A-H,O-Z)
IF (IOP .EQ. 0) GOTO 20
PRINT 10, N, ZA, ZB
10 FORMAT(1H1, 2X, 4HSTO-, I1,21HG FOR ATOMIC NUMBERS,
+ F5.2, 5H AND, F5.2)
CALL INTGRL(IOP, N, R, ZETA1, ZETA2, ZA, ZB) ! calculate integrals
CALL COLECT(IOP, N, R, ZETA1, ZETA2, ZA, ZB) ! put all integrals into arrays
CALL SCF(IOP, N, R, ZETA1, ZETA2, ZA, ZB) ! perform SCF calculation
20 CONTINUE
RETURN
ENDIntegral
SCF requires the evaluation of several integrals over the basis set $\{\phi_{\mu} \}$:
- $S_{\mu\nu}$
- $H_{\mu\nu}^{core} = T + \sum_{i}V_{i}$
- two-electron integral ($\mu \nu \mid \lambda \sigma$)
Overlap Integral
- overal integral has off-diagonal element calculated using equation (\ref{eq:1}):
- see equation 3.228 in chapter 3 of the book for the derivation
- this quantifies the degree to which the two atomic orbitals share the same space:
\begin{equation}
S_{\mu \nu} = \sum_{p=1}^{3} \sum_{q=1}^{L} d_{p\mu}^{*} d_{q\nu} S_{pq} \label{eq:1} \quad \mu, \nu = 1,2
\end{equation}
... S12 = S12 + S(A1(I), A2(J), R2)*D1(I)*D2(J) ...
- $S_{pq}$ is defined:
\begin{equation}
S_{pq} = \int d\mathbf{r} \phi_{p}^{GF}(\alpha_{p\mu}, r-R_A)\phi_{q}^{GF}(\alpha_{q\nu}, r-R_B)
\end{equation}
- this has analytical solution given in S(A,B,RAB2) function.
- final overlap matrix has the form: \(S=\left(\begin{array}{cc} 1 & S_{12} \\ S_{12} & 1 \end{array}\right)\)
Core Hamiltonian
Kinetic Energy Integral
- kinetic integral elements calculated using equation (\ref{eq:2}):
\begin{equation}
T_{\mu \nu} = \sum_{p=1}^{3} \sum_{q=1}^{L} d_{p\mu}^{*} d_{q\nu} T_{pq} \label{eq:2} \quad \mu, \nu = 1,2
\end{equation}
... T11 = T11 + T(A1(I), A1(J),0.0D0)*D1(I)*D1(J) T12 = T12 + T(A1(I), A2(J),R2)*D1(I)*D2(J) T22 = T22 + T(A2(I), A2(J),0.0D0)*D2(I)*D2(J) ... - $T_{pq}$ is defined:
\begin{equation}
T_{pq} = \int d\mathbf{r} \phi_{p}^{GF}(\alpha_{p\mu}, r-R_A)\left[ -\frac{1}{2}\nabla_r^2 \right]\phi_{q}^{GF}(\alpha_{q\nu}, r-R_B)
\end{equation}
- this has analytical solution calculated in T(A,B,RAB2) function based on equation \ref{eq:kinsolution}
- final kinetic energy matrix has the form, defining the core Hamiltonian matrix:
- represents the total energy of a single electron in the potential of the nuclei, before considering electron-electron repulsion \(T=\left(\begin{array}{cc} T_{11} & T_{12} \\ T_{12} & T_{22} \end{array}\right)\)
Potential Integral
-
general formulat for a potential energy integral, representing the attraction of an electron distribution to a nucleus C: \begin{equation} V_{\mu \nu, C} = \sum_{p=1}^{3} \sum_{q=1}^{L} d_{p\mu}^{*} d_{q\nu} V_{pq} \label{eq:3} \quad \mu, \nu = 1,2 \end{equation}
- attraction of an electron in one orbital to the other nucleus ($V_{12}, V_{21}$)
- attraction of an electron to its own nucleus ($V_{11},V_{22}$)
DO 20 I=1,N DO 20 J=1,N ... V11A = V11A + V(A1(I), A1(J), 0.0D0, 0.0D0, ZA)*D1(I)*D1(J) V12A = V12A + V(A1(I), A2(J), R2, RAP2, ZA)*D1(I)*D2(J) V22A = V22A + V(A2(I), A2(J), 0.0D0, R2, ZA)*D2(I)*D2(J) V11B = V11B + V(A1(I), A1(J), 0.0D0, R2, ZB)*D1(I)*D1(J) V12B = V12B + V(A1(I), A2(J), R2, RBP2, ZB)*D1(I)*D2(J) V22B = V22B + V(A2(I), A2(J), 0.0D0, 0.0D0, ZB)*D2(I)*D2(J) ... - $V_{pq}$ is defined:
\begin{equation}
V \rightarrow V_{pq} = \int d\mathbf{r} \phi_{p}^{GF}(\alpha_{p\mu}, r-R_A)\left[ -\frac{Z_c}{|r-R_C|} \right]\phi_{q}^{GF}(\alpha_{q\nu}, r-R_B)
\end{equation}
- solution of this is given in equation (\ref{eq:potsolution}), calculated using
V(A,B,RAB2,RCP2,ZC)function - these potential energy integrals are combined with the kinetic energy integrals to form the core Hamiltonian matrix with elements $H_{\mu\nu}^{core}$
- solution of this is given in equation (\ref{eq:potsolution}), calculated using
Two-electron Integral Derivation
- two-electron integrals are used for computing the $G$ matrix (see
FORMGsubroutine)- it accounts for the average repulsion an electron feels from all other electrons
- without this terms, the model incorrectly treat all electrons as independent particles, ignoring the fundamental repulsive forces between them
- a set of two-electron integrals (from equation 3.155 of the book): \begin{equation} (\mu\nu\mid\lambda\sigma) = \int d\mathbf{r_1}d\mathbf{r_2} \phi_{\mu}^{*}(1)\phi_{\nu}(1)\frac{1}{r_{12}} \phi_{\lambda}^{*}(2)\phi_{\sigma}(2) \label{eq:twoe} \end{equation}
- expanding this equation in the contracted Gaussian and evaluation of the resulting integral gives us:
\begin{equation}
(\mu \nu \mid \lambda \sigma)=\sum_{i=1}^N \sum_{j=1}^N \sum_{k=1}^N \sum_{l=1}^N d_{\mu, i} d_{\nu, j} d_{\lambda, k} d_{\sigma, l}\left(g_i g_j \mid g_k g_l\right)
\end{equation}
- computational equivalent:
DO 30 I=1,N DO 30 J=1,N DO 30 K=1,N DO 30 L=1,N ... V1111 = V1111 + TWOE(A1(I),A1(J),A1(K),A1(L),0.0D0,0.0D0,0.0D0) + *D1(I)*D1(J)*D1(K)*D1(L) V2111 = V2111 + TWOE(A2(I),A1(J),A1(K),A1(L),R2,0.0D0,RAP2) + *D2(I)*D1(J)*D1(K)*D1(L) V2121 = V2121 + TWOE(A2(I),A1(J),A2(K),A1(L),R2,R2,RPQ2) + *D2(I)*D1(J)*D2(K)*D1(L) V2211 = V2211 + TWOE(A2(I),A2(J),A1(K),A1(L),0.0D0,0.0D0,R2) + *D2(I)*D2(J)*D1(K)*D1(L) V2221 = V2221 + TWOE(A2(I),A2(J),A2(K),A1(L),0.0D0,R2,RBQ2) + *D2(I)*D2(J)*D2(K)*D1(L) V2222 = V2222 + TWOE(A2(I),A2(J),A2(K),A2(L),0.0D0,0.0D0,0.0D0) + *D2(I)*D2(J)*D2(K)*D2(L) ... - $g_i$ is the primitive Gaussians where solution of such integrals are derived in Appendix A of the book
- computation is given by the
TWOE(A, B, C, D, RAB2, RCD2, RPQ2)function
- computational equivalent:
Electron Repulsion G matrix
- after obtaining the core Hamiltonina, next is to calculate the two-electron part of the Fock Matrix $G_{\mu\nu}$
\begin{equation}
F_{\mu\nu } = H_{\mu\nu}^{core} + G_{\mu\nu}
\end{equation}
- depends on the density matrix $\mathbf{P}$ and the set of two-electron integrals $(\mu\nu\mid\lambda\sigma)$ (see equation \ref{eq:twoe})
-
$\mathbf{G}$ matrix: \(\begin{equation} \begin{split} G_{\mu\nu} &= 2\sum_{a}^{N/2} \sum_{\lambda \sigma} C_{\lambda a}C_{\sigma a}^{*}[(\mu\nu\mid\sigma\lambda) - \frac{1}{2} (\mu\lambda\mid\sigma\nu)] \\ &= \sum_{\lambda \sigma} P_{\lambda \sigma}[(\mu\nu\mid\sigma\lambda) - \frac{1}{2} (\mu\lambda\mid\sigma\nu)] \end{split} \label{eq:gmatrix} \end{equation}\)
DO 10, K=1,2 DO 10, L=1,2 G(I,J) = G(I,J) + P(K,L)*(TT(I,J,K,L)-0.5D0*TT(I,L,K,J))
- density matrix/charge-density bond-order matrix is defined as:
\begin{equation}
P_{\lambda \sigma} = 2\sum_{a}^{N/2} C_{\lambda a}C_{\sigma a}^{*} \label{eq:densitymat}
\end{equation}
- how do we compute the density matrix $\mathbf{P}$??
- first is an initial guess, then use SCF procedure to update its value
- how do we compute the density matrix $\mathbf{P}$??
Collecting the elements
COLECTsubroutine purpose is to form the relevant matrices for SCF:- $\mathbf{S}, \mathbf{H}, \mathbf{X}, X^{\dagger}$, and two-electron integral $(\mu\nu\mid\lambda\sigma)$
- overlap matrix, $\mathbf{S}$:
S(1,1) = 1.0D0 S(1,2) = S12 S(2,1) = S(1,2) S(2,2) = 1.0D0 - core Hamiltonian, $\mathbf{H}$:
H(1,1) = T11 + V11A + V11B H(1,2) = T12 + V12A + V12B H(2,1) = T12 + V12A + V12B H(2,2) = T22 + V22A + V22B - transformation matrix, $\mathbf{X}$, $\mathbf{X^{\dagger}}$:
! use canonical orthogonalization to form the X matrix X(1,1) = 1.0D0/DSQRT(2.0D0*(1.0D0 + S12)) X(1,2) = X(1,1) X(2,1) = 1.0D0/DSQRT(2.0D0*(1.0D0 - S12)) X(2,2) = -X(1, 2) ! form the X transpose matrix XT(1,1) = X(1,1) XT(1,2) = X(2,1) XT(2,1) = X(1,2) XT(2,2) = X(2,2) - two-electron integral matrix, $(\mu\nu\mid\lambda\sigma)$:
TT(1,1,1,1) = V1111 TT(2,1,1,1) = V2111 TT(1,2,1,1) = V2111 TT(1,1,2,1) = V2111 TT(1,1,1,2) = V2111 TT(2,1,2,1) = V2121 TT(1,2,2,1) = V2121 TT(2,1,1,2) = V2121 TT(1,2,1,2) = V2121 TT(2,2,1,1) = V2211 TT(1,1,2,2) = V2211 TT(2,2,2,1) = V2221 TT(2,2,1,2) = V2221 TT(2,1,2,2) = V2221 TT(1,2,2,2) = V2221 TT(2,2,2,2) = V2222
Self-Consistent Field (SCF) Procedure
- algorithm of the SCF (page 146 of the book), code snippet are from
SCFsubroutine.- diagonalize the overlap matrix S and obtain a transformation matrix X from symmetric orthogonalization: $\mathbf{X} \equiv \mathbf{S}^{-1/2} $
- obtain a guess at the density matrix $\mathbf{P}$
- calculate the matrix $\mathbf{G}$ of equation (\ref{eq:gmatrix}) from the density matrix $P$ and the two-electron integrals $(\mu\nu\mid\lambda\sigma)$
- Add $\mathbf{G}$ to the core-Hamiltonian to obtain the Fock matrix $\mathbf{F}=\mathbf{H}^{core} + \mathbf{G}$
DO 60 I=1,2 DO 60 J=1,2 F(I,J) = H(I,J) + G(I,J) - Calculate the transformed matrix $\mathbf{F’}=\mathbf{X^{\dagger}FX}$
CALL MULT(F,X,G,2,2) CALL MULT(XT,G,FPRIME,2,2) - Diagonalize $\mathbf{F’}$ to obtain $\mathbf{C’}$ and $\epsilon$
CALL DIAG(FPRIME,CPRIME,E) - Calculate $\mathbf{C=XC’}$
CALL MULT(X,CPRIME,C,2,2) - form a new density matrix $\mathbf{P}_{new}$ from $\mathbf{C}$ using equation (\ref{eq:densitymat}): \(\mathbf{P}\rightarrow \mathbf{P}_{old}, \mathbf{P}_{new} \rightarrow \mathbf{P}\)
DO 100 I=1,2 DO 100 J=1,2 OLDP(I,J) = P(I,J) P(I,J) = 0.0D0 DO 100 K=1, 1 P(I,J) = P(I,J) + 2.0D0*C(I,K)*C(J,K) - determined whether the procedure has converged: $|\mathbf{P}-\mathbf{P}_{old}| < \text{CRIT} $
DATA CRIT/1.0D-4/ ... DO 120 I=1,2 DO 120 J=1,2 DELTA = DELTA + (P(I,J) - OLDP(I,J))**2 120 CONTINUE DELTA = DSQRT(DELTA/4.0D0) ... IF (DELTA .LT. CRIT) GO TO 160 IF (ITER .LT. MAXIT) GO TO 20 - if it has converged, use the resulting solution ($\mathbf{C, P, F,..}$) to calculate the expectation values and other quantities of interest
... 160 CONTINUE ! add nuclear repulsion energy to get the total energy ENT = EN + ZA*ZB/R ...- other expectation values: energy, dipole moment, etc.
- population analyses
Integral Solutions
Two-Electron Integral
FUNCTION TWOE(A, B, C, D, RAB2, RCD2, RPQ2)
IMPLICIT DOUBLE PRECISION (A-H,O-Z)
DATA PI/3.1415926535898D0/
TWOE=2.0D0*PI**(2.5D0)/((A+B)*(C+D)*DSQRT(A+B+C+D))
+ *F0((A+B)*(C+D)*RPQ2/(A*B+C*D))
+ *DEXP(-A*B*RAB2/(A+B)-C*D*RCD2/(C+D))
RETURN
END where $ T = \frac{(a+b)(c+d)}{a+b+c+d}R^2_{PQ}$ and $F_0(T)$ is the error function computed using F0(ARG) function.
Overlap Energy
- see Appendix A for the derivation: \begin{equation} \left(A | B \right) = \frac{\pi}{(\alpha + \beta)}^{3/2} \exp\left[\frac{-\alpha\beta}{(\alpha + \beta)}|R_{AB}|^2\right] \end{equation} where: $R_{AB} = |R_A -R_B|$
FUNCTION S(A,B,RAB2)
IMPLICIT DOUBLE PRECISION(A-H, O-Z)
DATA PI/3.1415926535898D0/
S = (PI/(A+B))**1.5D0*DEXP(-A*B*RAB2/(A+B))
RETURN
ENDKinetic Energy
- see Appendix A for the derivation: \begin{align} \left(A\left|-\frac{1}{2}\nabla^2\right|B\right) = \frac{\alpha \beta}{(\alpha + \beta)} \ [3 - \frac{2\alpha\beta}{(\alpha + \beta)}|R_{AB}|^2]\left[\frac{\pi}{(\alpha + \beta)}\right]^{3/2}\exp\left[\frac{-\alpha\beta}{(\alpha + \beta)}|R_{AB}|^2\right] \label{eq:kinsolution} \end{align}
FUNCTION T(A,B,RAB2)
IMPLICIT DOUBLE PRECISION(A-H, O-Z)
DATA PI/3.1415926535898D0/
T=A*B/(A+B)*(3.0D0-2.0D0*A*B*RAB2/(A+B))*(PI/(A+B))**1.5D0
+ *DEXP(-A*B*RAB2/(A+B))
RETURN
ENDPotential Energy
- see Appendix A for the derivation: \begin{equation} \left(A\left|-\frac{Z_C}{r_{1C}}\right|B\right) = \frac{-2\pi}{(\alpha + \beta)}Z_c \exp\left[ \frac{-\alpha \beta}{(\alpha + beta)}|R_A - R_B|^2\right] \times F_0[(\alpha + \beta)|R_p - R_C|^2] \label{eq:potsolution} \end{equation}
FUNCTION V(A,B,RAB2,RCP2,ZC)
IMPLICIT DOUBLE PRECISION(A-H, O-Z)
DATA PI/3.1415926535898D0/
V=2.0D0*PI/(A+B)*FO((A+B)*RCP2)*DEXP(-A*B*RAB2/(A+B))
V = -V*ZC
RETURN
ENDDiagonalization
- subroutine uses the Jacobi rotation method, which for a 2x2 matrix provides an exact, analytical solution. \begin{equation} FC = CE \end{equation}
- constructing the eigenvectors $C$
- finding the rotation matrix $\theta$:
- computing the eigenvalues, which are the diagonal elements of the transformed matrix $E=C^TFC$, code uses the explicit formulas for these elements:
SUBROUTINE DIAG(F,C,E)
IMPLICIT DOUBLE PRECISION(A-H,O-Z)
DIMENSION F(2,2), C(2,2), E(2,2)
DATA PI/3.1415926535898D0/
IF (DABS(F(1,1)-F(2,2)) .GT. 1.0D-20) GO TO 10
THETA = PI/4.0D0
GO TO 20
10 CONTINUE
THETA = 0.5D0*DATAN(2.0D0*F(1,2)/(F(1,1)-F(2,2)))
20 CONTINUE
C(1,1) = DCOS(THETA)
C(2,1) = DSIN(THETA)
C(1,2) = DSIN(THETA)
C(2,2) = -DCOS(THETA)
E(1,1) = F(1,1)*DCOS(THETA)**2+F(2,2)*DSIN(THETA)**2
$ +F(1,2)*DSIN(2.0D0*THETA)
E(2,2) = F(2,2)*DCOS(THETA)**2+F(1,1)*DSIN(THETA)**2
$ -F(1,2)*DSIN(2.0D0*THETA)
E(2,1) = 0.0D0
E(1,2) = 0.0D0
IF(E(2,2) .GT. E(1,1)) GO TO 30
TEMP = E(2,2)
E(2,2) = E(1,1)
E(1,1) = TEMP
TEMP = C(1,2)
C(1,2) = C(1,1)
C(1,1) = TEMP
TEMP = C(2,2)
C(2,2) = C(2,1)
C(2,1) = TEMP
30 RETURN
ENDAuxilliary Subroutine, Function
Matrix Multiplication
SUBROUTINE MULT(A,B,C,IM,M)
IMPLICIT DOUBLE PRECISION(A-H,O-Z)
DIMENSION A(IM,IM), B(IM,IM), C(IM,IM)
DO 10, I=1,M
DO 10, J=1,M
C(I,J)=0.0D0
DO 10 K=1,M
10 C(I,J) = C(I,J) + A(I,K)*B(K,J)
RETURN
ENDError Function
FUNCTION FO(ARG)
IMPLICIT DOUBLE PRECISION(A-H,O-Z)
DATA PI/3.1415926535898D0/
IF (ARG .LT. 1.0D-6) GO TO 10
FO=DSQRT(PI/ARG)*DARF(DSQRT(ARG))/2.0D0
GO TO 20
10 FO=1.0D0-ARG/3.0D0
20 CONTINUE
RETURN
END FUNCTION DARF(ARG)
IMPLICIT DOUBLE PRECISION(A-H,O-Z)
DIMENSION A(5)
DATA P/0.3275911D0/
DATA A/0.254829592D0, -0.284496736D0,1.421413741D0,
$ -1.453152027D0,1.061405429D0/
T=1.0D0/(1.0D0+P*ARG)
TN = T
POLY = A(1)*TN
DO 10 I=2,5
TN = TN*T
POLY=POLY + A(I)*TN
10 CONTINUE
DARF = 1.0D0-POLY*DEXP(-ARG*ARG)
RETURN
END